Skin in Genetic and Metabolic Disease

Many genetic and metabolic diseases are associated with changes in the skin. In some conditions, such as phenylketonuria (PKU), a skin rash may be the first sign of the disease. Being able to recognize the cutaneous manifestations of genetic and metabolic disease will provide immediate visual clues to the diagnosis. While genetic testing continues to advance, it is still possible to obtain a wealth of information just by looking at the patient.
Vitamin Deficiencies
Vitamin B3 (Niacin)
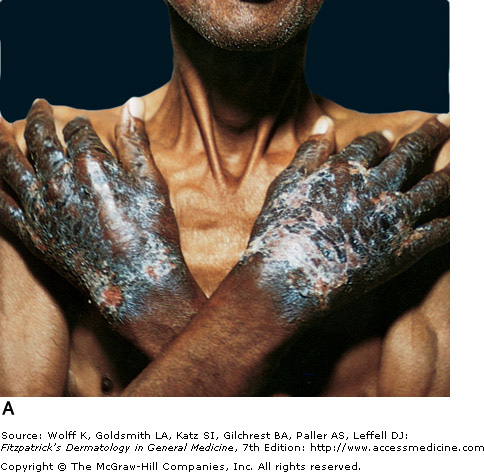
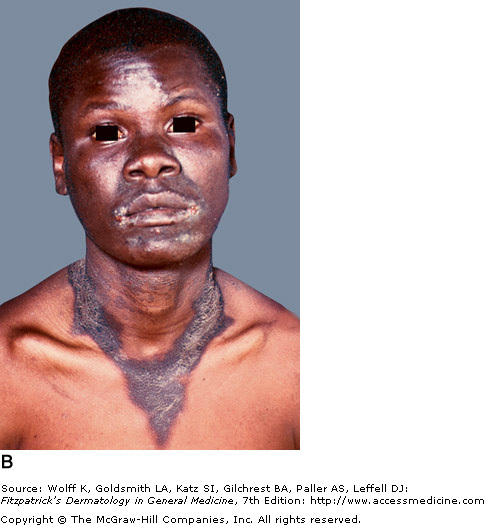
Vitamin B3 is obtained in the diet or synthesized in the body from tryptophan. A corn rich diet is problematic with niacin's main source in foods such as whole grains, dairy, beans, and meat. A deficiency in B3 causes pellagra, which is known by its "Four Ds" (dermatitis, diarrhea, dementia, and death). The dermatitis is caused by photosensitivity, initially presenting as erythematous, painful, or itchy patches. It further develops edema with vesicles and bullae, which upon rupture leave crusted erosions. The skin eventually thickens and manifests hyperpigmented plaques that are sharply demarcated. Sun exposed areas, such as the hands and face, are most affected. Casal's necklace is the name given for the around the neck. Treatment consists of supplemental niacin or tryptophan. Neurological symptoms can improve within days of treatment, but the skin will take 3-4 weeks to heal.
Zinc
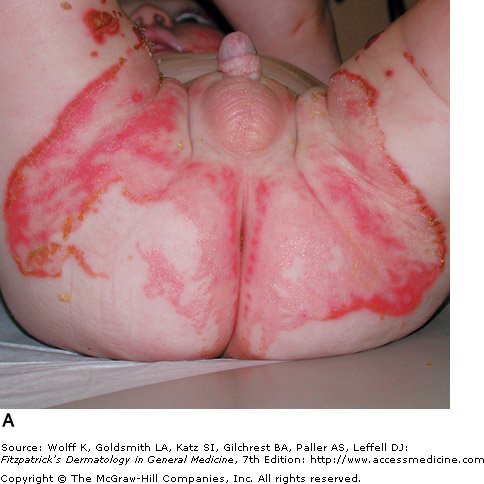
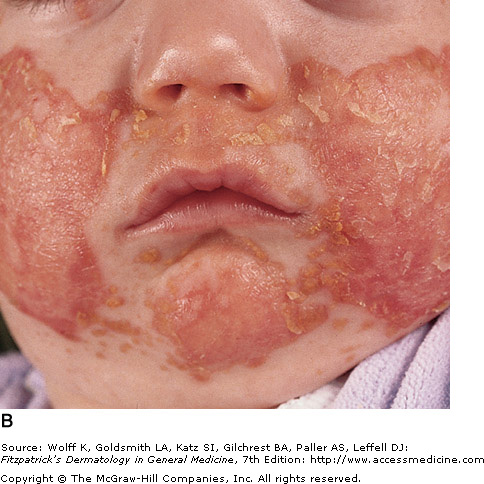
Zinc deficiency can be either inherited or acquired. Acrodermatitis enteropathica (AE) is caused by an inherited zinc deficiency, classically presenting when an infant stops breast feeding. Common symptoms include alopecia (hair loss), diarrhea, lethargy, and eczematous/erosive dermatitis in the perioral, periocular, anogenital, hands, and feet areas. Eventually, bullae and erosions with a characteristic crusted border will develop. Zinc deficient patients have a higher predisposition to infections, as their cell-mediated immunity is compromised. Infection with Candida albicans and Staphylococcus aureus are common superinfections that may occur in these patients. Treatment of AE with zinc supplementation usually responds rapidly, and patients improve in a few days.
Vitamin B3 is obtained in the diet or synthesized in the body from tryptophan. A corn rich diet is problematic with niacin's main source in foods such as whole grains, dairy, beans, and meat. A deficiency in B3 causes pellagra, which is known by its "Four Ds" (dermatitis, diarrhea, dementia, and death). The dermatitis is caused by photosensitivity, initially presenting as erythematous, painful, or itchy patches. It further develops edema with vesicles and bullae, which upon rupture leave crusted erosions. The skin eventually thickens and manifests hyperpigmented plaques that are sharply demarcated. Sun exposed areas, such as the hands and face, are most affected. Casal's necklace is the name given for the around the neck. Treatment consists of supplemental niacin or tryptophan. Neurological symptoms can improve within days of treatment, but the skin will take 3-4 weeks to heal.
Zinc
Zinc deficiency can be either inherited or acquired. Acrodermatitis enteropathica (AE) is caused by an inherited zinc deficiency, classically presenting when an infant stops breast feeding. Common symptoms include alopecia (hair loss), diarrhea, lethargy, and eczematous/erosive dermatitis in the perioral, periocular, anogenital, hands, and feet areas. Eventually, bullae and erosions with a characteristic crusted border will develop. Zinc deficient patients have a higher predisposition to infections, as their cell-mediated immunity is compromised. Infection with Candida albicans and Staphylococcus aureus are common superinfections that may occur in these patients. Treatment of AE with zinc supplementation usually responds rapidly, and patients improve in a few days.
|
|
Alkaptonuria
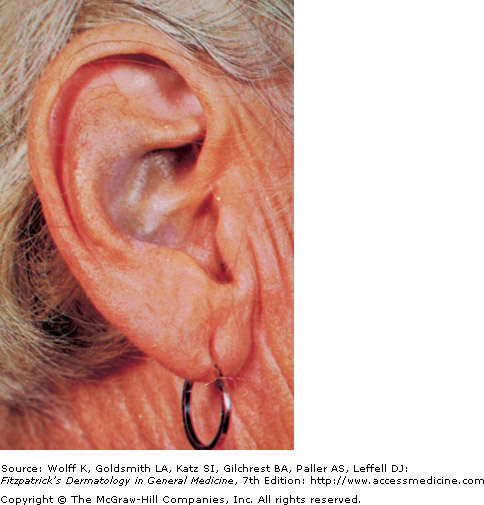
Discoloration of Ear Cartilage
Also known as ochronosis, this homogentisic acid oxidase deficiency is a rare metabolic disorder in which homogentisic acid builds up in excess in the body's connective tissues, including the dermis. This autosomal recessive disorder is marked by excess homogentisic acid in the urine, which turns dark in color which may show as a brownish discoloration of the diaper. Although it may be diagnosed in childhood, it is typically not diagnosed until the third or fourth decade of life, when connective tissue problems are noticed.
The primary clinical features include blue-black or gray discoloration of sclerae, face, pinnae, cartilage, and nails though axillary discoloration may be present in the teen years. Arthritis is also common, as is dark colored sweat. Alkaptonuria is typically slow and irreversible, and only supportive treatment exists. High amounts of vitamin C can be usful in protecting homogentisic acid against oxidation, preventing the deposition of pigment. Diets avoiding high amounts of protein, phenylalamine, and tyrosine are recommended.
The primary clinical features include blue-black or gray discoloration of sclerae, face, pinnae, cartilage, and nails though axillary discoloration may be present in the teen years. Arthritis is also common, as is dark colored sweat. Alkaptonuria is typically slow and irreversible, and only supportive treatment exists. High amounts of vitamin C can be usful in protecting homogentisic acid against oxidation, preventing the deposition of pigment. Diets avoiding high amounts of protein, phenylalamine, and tyrosine are recommended.
Amyloidosis
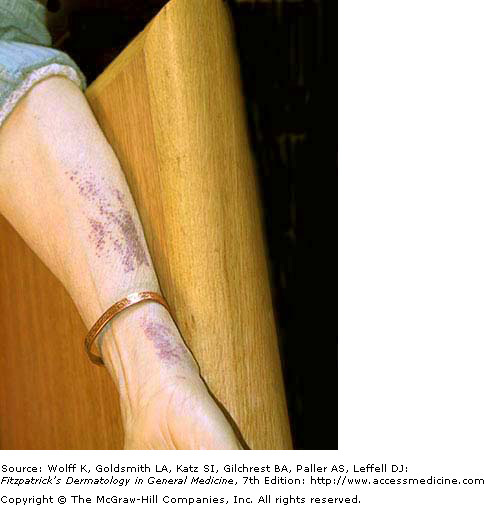
Purpuric Rash - Hereditary Amyloidosis
Amyloidosis refers to a family of conditions in which amyloid proteins (insoluble fibrous protein aggregates) are abnormally deposited into tissues. When the skin is involved in the deposition of these proteins, a variety of problems can occur, such as hyperpigmented keratotic lesions, purpura, and xanthomatous papules and plaques. In hereditary amyloidosis, a liver transplant is often needed in order to reduce the production of these rogue amyloid proteins. Organs that have excess protein deposition may also fail and will require transplantation. Median survival with a liver transplant is 10-15 years. Systemic AA amyloidosis occurs without a primary hereditary component, and is seen with chronic inflammatory diseases, such as rheumatoid arthritis. This disease is characterized by proteinuria and eventual renal failure.
Images below: Be sure to note that lesions can range from papules to plaques. On histological examination, the dermis appears separated due to excessive protein deposits.
Images below: Be sure to note that lesions can range from papules to plaques. On histological examination, the dermis appears separated due to excessive protein deposits.
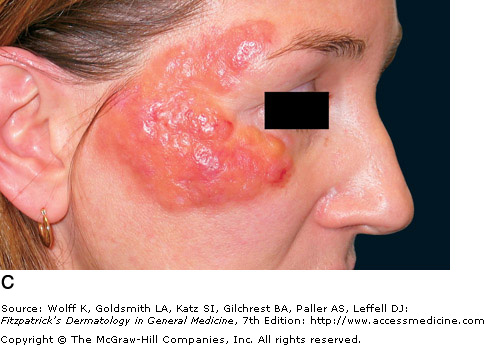 Amyloidosis Plaque - Fitzpatrick's Dermatology |
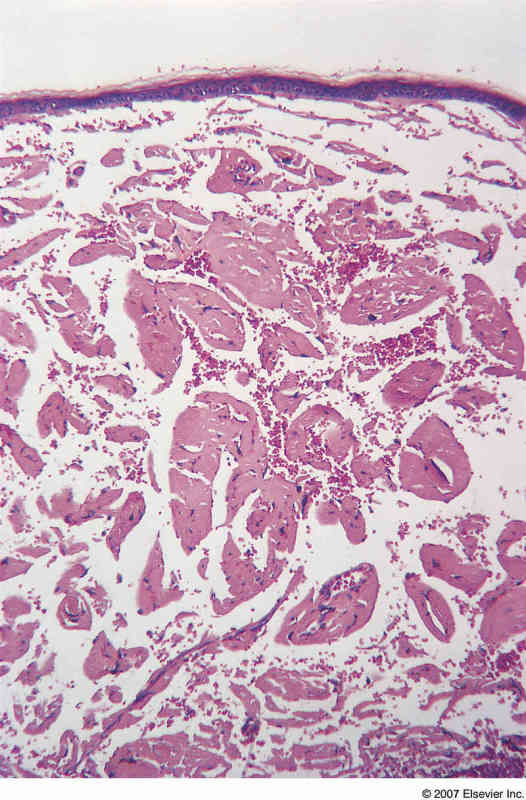 Amyloidosis Histopathology - PathConsult |
Xeroderma Pigmentosum
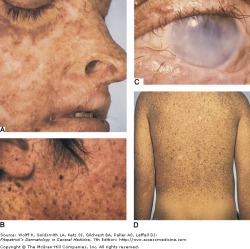
Xeroderma Pigmentosum - Fitzpatrick's Dermatology
Xeroderma pigmentosum (XP) is a rare autosomal recessive disease of increased sensitivity to cellular injury due to defects in DNA repair. This results in sun sensitivity, photophobia, freckling, and skin cancers. This disease is quite rare (1 case per 1 million people in the US), but heterozygote carriers may be at increased skin cancer risk.
A large amount of freckling occurs on sun-exposed skin, and with continuous sun exposure the skin becomes dry and parchment-like. The median age of onset of XP skin symptoms is 1-2 years old. XP patients have a more than 1000x increased risk for skin cancers, with a non-melanoma skin cancer occuring at a median age of 8 years. Patients are also at a greater risk for central nervous tumors and very often have ocular and neurologic complications. A defect in the necleotide excision repair (NER) pathway of DNA after ultraviolet light damage is responsible for the underlying neoplasms and other complications of XP. Management consists of rigorous sun protection, including long clothing, UV-absorbing glasses, and sunscreen.
A large amount of freckling occurs on sun-exposed skin, and with continuous sun exposure the skin becomes dry and parchment-like. The median age of onset of XP skin symptoms is 1-2 years old. XP patients have a more than 1000x increased risk for skin cancers, with a non-melanoma skin cancer occuring at a median age of 8 years. Patients are also at a greater risk for central nervous tumors and very often have ocular and neurologic complications. A defect in the necleotide excision repair (NER) pathway of DNA after ultraviolet light damage is responsible for the underlying neoplasms and other complications of XP. Management consists of rigorous sun protection, including long clothing, UV-absorbing glasses, and sunscreen.
